您现在的位置是:首页 >学无止境 >C#,生信软件实践(05)——序列比对格式SAM/BAM文件的详解介绍及解释器之完整C#源代码网站首页学无止境
C#,生信软件实践(05)——序列比对格式SAM/BAM文件的详解介绍及解释器之完整C#源代码
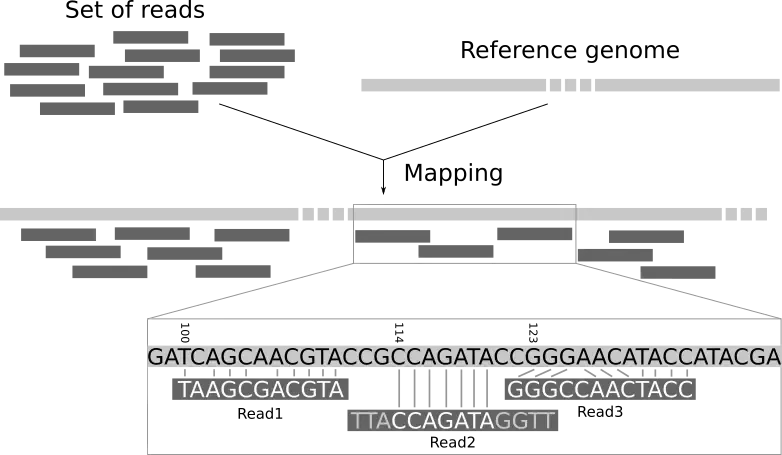
SAM/BAM/CRAM Format
The official SAM documentation can be found here.
These formats were introduced to standardize how alignments are reported. Initially there were many different formats, most of them proprietary, which were space inefficient and either held too much or too little information. The first of these to be introduced was Sequence Alignment Map (SAM). With this format not only is the alignment retained but the associated quality scores (both mapping and base quality), the original read itself, paired-end information, sample information, and many more features.
SAM Format
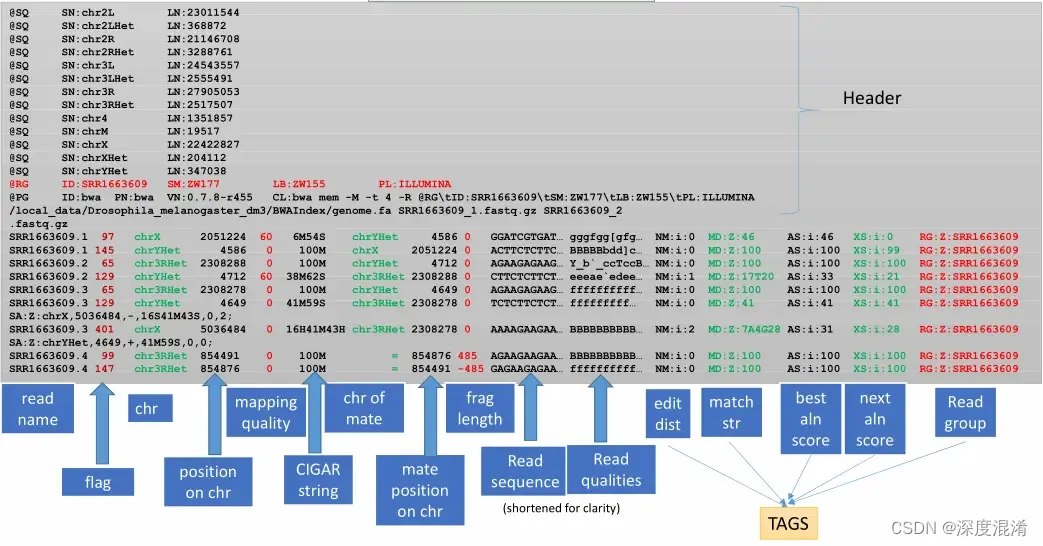
This is the most basic, human readable format of the three. This is generated by almost every alignment algorithm that exists. It consists of a header, a row for every read in your dataset, and 11 tab-delimited fields describing that read.
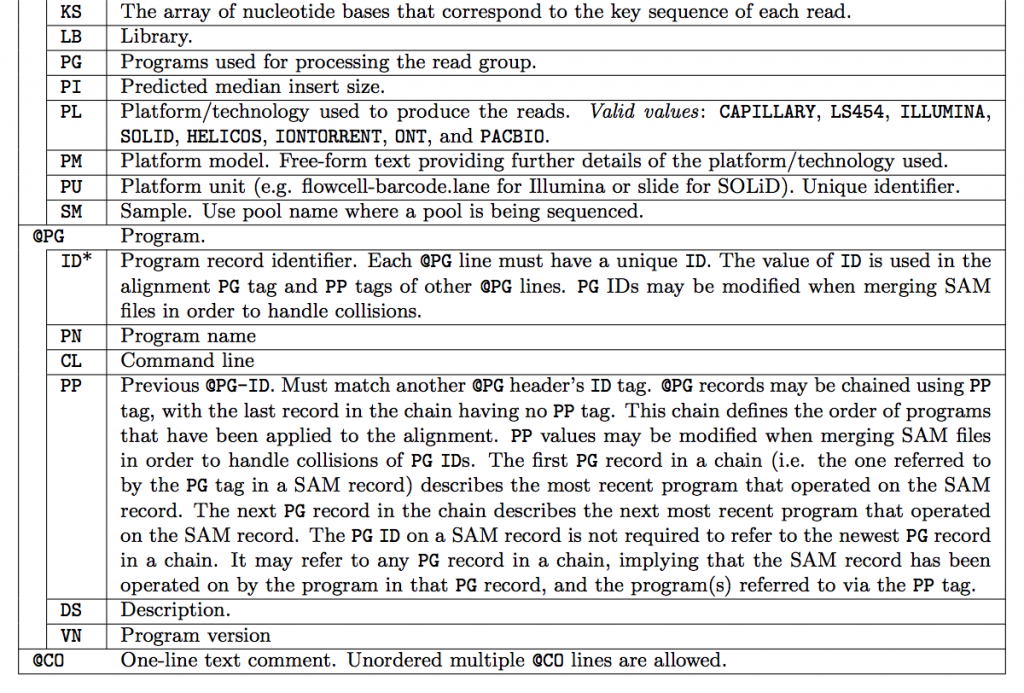
SAM Header
The header varies in size but adheres to a particular format depending on what information you decide to add. Some example information that can be entered into the header is: command that generated the SAM file, SAM format version, sequencer name and version. The full list of available header fields can be found below.
http://samtools.github.io/hts-specs/SAMv1.pdf
http://samtools.github.io/hts-specs/SAMv1.pdf
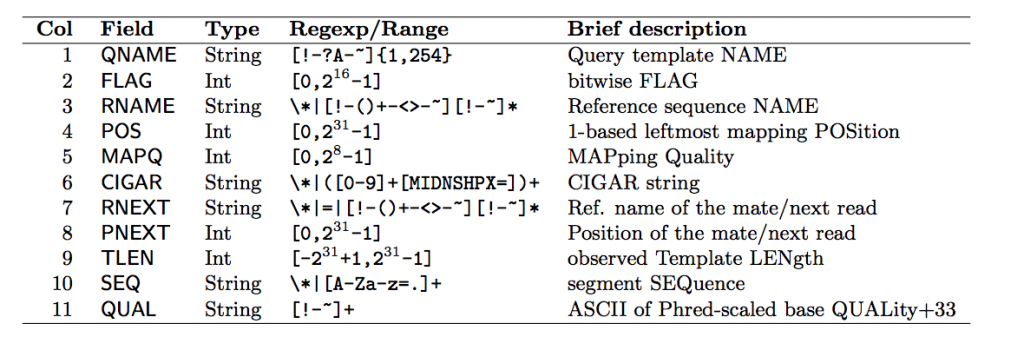
Field Descriptions
Each row contains 11 mandatory fields. The descriptions for them can be found below:
http://samtools.github.io/hts-specs/SAMv1.pdf
Let’s look at some of the fields that aren’t very self explanatory:
Bitwise Flag
The bitwise flag is a lookup code to explain certain features about the particular read (exact same concept as Linux permission codes!). It tells you whether the read aligned, is marked a PCR duplicate, if it’s mate aligned, etc. and any combination of the available tags, seen below:

http://samtools.github.io/hts-specs/SAMv1.pdf
One important thing to note is that any combination of these flags results in one integer, which makes interpreting it a bit difficult. To make it easy you can check here to either encode or decode a bitwise flag.
MapQ (Mapping Quality)
This value reports how well the read aligned to the reference. Different algorithms report it differently but nonetheless, the greater the number the better the alignment (generally).
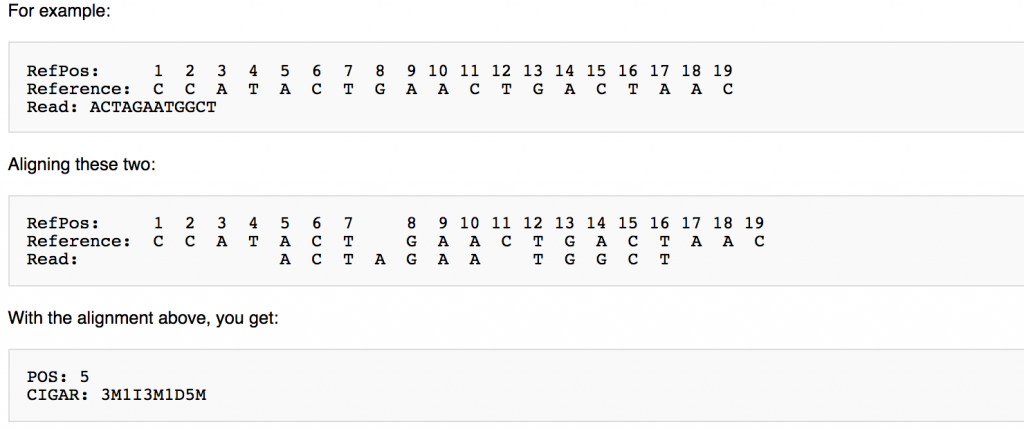
CIGAR String
This is a shorthand way to encode an entire alignment. Instead of writing the whole alignment out, operators have been defined and are used in combination with numbers to explain which part of the sequence aligns, which doesn’t, and everything in between. The definition for the operators can be found here:

https://www.drive5.com/usearch/manual/cigar.html
https://genome.sph.umich.edu/wiki/SAM#What_is_a_CIGAR.3F
Example SAM file
HWI-ST865:416:C6CG0ACXX:1:1313:9073:43827 0 I 2 1 99M * 0 0 CCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT @C@DFDEFFHDFFIIJIGIIGIGGIIIIJGHGIJJEEIAHHGGIGFH@HGCFGGGJJIIGDAFG@DGIHHHHHFFBB@CACEC6;?CDD?CDCAD>>AA AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:99 YT:Z:UU
HWI-ST865:416:C6CG0ACXX:1:1215:16359:6484 16 I 9 1 85M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCC EEEEFFFFDAGHHHIJJIIJJJJJIJJJJIJIJJIGIIJJJJJJJIJJJJJJJIIJJJIIJGJJIJJJJJJJHFHHHFFFFFCCB AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:85 YT:Z:UU
HWI-ST865:416:C6CG0ACXX:1:1113:14118:89232 16 I 15 1 100M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAA CAC@A>C@AADCAC@ACEEC@@BD?E;@CEHGCEIGIHAFHGGF;FCHBHFBHIGIIIJJJJJJJJJJJJJJJJJJJJJIJJJJJJJHHHHHFFFFFCCC AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU
HWI-ST865:412:C6CLLACXX:1:2315:12173:84819 16 I 49 1 70M * 0 0 GCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT @7)3CC=)CA;EBC>DAEDBDCDDDCDD@B?<DEDE399CBC<+>EAE<BDCEAE3DADDD<ABDD;1?? AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:70 YT:Z:UU HWI-ST865:412:C6CLLACXX:1:1201:19323:33842 16 I 71 0 100M * 0 0 AAGCCTAAGCCTAAGCCTAAGCCTAAGCCAAATCCCAAGCCTAAGCCTAAGCCTAAGCCTAAGCCAGAGCCTAAGCCTAAGCCTTAGCCTAAGCCTGATC DDDCCDCCACCCDCCCC>CAA@D@;BDHA3A7(@5/IIHFIIGEIIIIHEIIIIIGGIGIIIIIFDCIIIIIIIIGGIIGIHFFEIHDDBHFFDDDB@@@ AS:i:-33 XS:i:-33 XN:i:0 XM:i:8 XO:i:0 XG:i:0 NM:i:8 MD:Z:29T2G2T29T0A17A11A1G1 YT:Z:UU
HWI-ST865:412:C6CLLACXX:1:1215:19021:78287 16 I 93 1 66M * 0 0 CTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGC @=87=3GF=.:CFE;D@B3?3?BD9BC<>CJJJJJJJJJJJJJJJJIJJJJJIHHHHHFFEDAC@B AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:66 YT:Z:UU
BAM Format
This is the same format except that it encoded in binary which means that it is significantly smaller than the SAM files and significantly faster to read, though it is not human legible and needs to be converted to another format (i.e. SAM) in order to make sense to us.
Some special tools are needed in order to make sense of BAM, such as Samtools, Picard Tools, and IGV which will be discussed in some of the latter sections.
using System;
using System.IO;
using System.Text;
using System.Collections;
using System.Collections.Generic;
using System.Runtime.Serialization;
namespace Legal.BIOG
{
/// <summary>
/// SAM对比数据
/// @HD VN:1.0 SO:coordinate
/// @SQ SN:chr1 LN:249250621
/// @SQ SN:chr10 LN:135534747
/// @SQ SN:chr11 LN:135006516
/// ...
/// @SQ SN:chrY LN:59373566
/// @PG ID:TopHat VN:2.0.8b CL:/home/hpages/tophat-2.0.8b.Linux_x86_64/tophat --mate-inner-dist 150 --solexa-quals --max-multihits 5 --no-discordant --no-mixed --coverage-search --microexon-search --library-type fr-unstranded --num-threads 2 --output-dir tophat2_out/ERR127306 /home/hpages/bowtie2-2.1.0/indexes/hg19 fastq/ERR127306_1.fastq fastq/ERR127306_2.fastq
/// https://www.samformat.info/sam-format-header
/// </summary>
public class SAM_Head
{
/// <summary>
/// One-line text comment
/// Unordered multiple @CO lines are allowed
/// </summary>
public string CO { get; set; } = "";
/// <summary>
/// The header line
/// The first line if present.
/// 说明VN的版本以及比对有无排列顺序,这个例子没有排序。
/// </summary>
public string HD { get; set; } = "";
/// <summary>
/// Program
/// 使用的比对程序名。
/// </summary>
public string PG { get; set; } = "";
/// <summary>
/// Read group
/// Unordered multiple @RG lines are allowed.
/// </summary>
public List<string> RG { get; set; } = new List<string>();
/// <summary>
/// Reference sequence dictionary
/// The order of @SQ lines defines the alignment sorting order.
/// 参考序列目录。SN:参考序列名字。LN:参考序列长度。
/// </summary>
public List<string> SQ { get; set; } = new List<string>();
public SAM_Head() { }
/// <summary>
/// https://samtools.github.io/hts-specs/SAMv1.pdf
/// </summary>
public string HD_Details
{
get
{
List<string> ha = B.S2T(HD);
StringBuilder sb = new StringBuilder();
foreach (string hx in ha)
{
if (hx.StartsWith("VN:")) sb.AppendLine("format version: " + hx.Substring(3));
else if (hx.StartsWith("SO:")) sb.AppendLine("Sorting order of alignments: " + hx.Substring(3));
else if (hx.StartsWith("GO:")) sb.AppendLine("Grouping of alignments: " + hx.Substring(3));
else if (hx.StartsWith("SS:")) sb.AppendLine("Sub-sorting order of alignments: " + hx.Substring(3));
else sb.AppendLine("Undefined: " + hx);
}
return sb.ToString();
}
}
public string PG_Details
{
get
{
List<string> ha = B.S2T(PG);
StringBuilder sb = new StringBuilder();
foreach (string hx in ha)
{
if (hx.StartsWith("ID:")) sb.AppendLine("Program record identifier: " + hx.Substring(3));
else if (hx.StartsWith("PN:")) sb.AppendLine("Program name: " + hx.Substring(3));
else if (hx.StartsWith("CL:")) sb.AppendLine("Command line: " + hx.Substring(3));
else if (hx.StartsWith("PP:")) sb.AppendLine("Previous @PG-ID: " + hx.Substring(3));
else if (hx.StartsWith("DS:")) sb.AppendLine("Description: " + hx.Substring(3));
else if (hx.StartsWith("VN:")) sb.AppendLine("Program version: " + hx.Substring(3));
else sb.AppendLine("Undefined: " + hx);
}
return sb.ToString();
}
}
public string RG_Details
{
get
{
StringBuilder sb = new StringBuilder();
foreach (string rx in RG)
{
List<string> ha = B.S2T(rx);
foreach (string hx in ha)
{
if (hx.StartsWith("ID:")) sb.AppendLine("Read group identifier: " + hx.Substring(3));
else if (hx.StartsWith("BC:")) sb.AppendLine("Barcode sequence identifying the sample or library: " + hx.Substring(3));
else if (hx.StartsWith("CN:")) sb.AppendLine("Name of sequencing center producing the read: " + hx.Substring(3));
else if (hx.StartsWith("DS:")) sb.AppendLine("Description: " + hx.Substring(3));
else if (hx.StartsWith("DT:")) sb.AppendLine("Date the run was produced (ISO8601 date or date/time): " + hx.Substring(3));
else if (hx.StartsWith("FO:")) sb.AppendLine("Flow order: " + hx.Substring(3));
else if (hx.StartsWith("KS:")) sb.AppendLine("The array of nucleotide bases that correspond to the key sequence of each read: " + hx.Substring(3));
else if (hx.StartsWith("LB:")) sb.AppendLine("Library: " + hx.Substring(3));
else if (hx.StartsWith("PG:")) sb.AppendLine("Programs used for processing the read group: " + hx.Substring(3));
else if (hx.StartsWith("PI:")) sb.AppendLine("Predicted median insert size: " + hx.Substring(3));
else if (hx.StartsWith("PL:")) sb.AppendLine("Platform/technology used to produce the reads: " + hx.Substring(3));
else if (hx.StartsWith("PM:")) sb.AppendLine("Platform model: " + hx.Substring(3));
else if (hx.StartsWith("PU:")) sb.AppendLine("Platform unit, a unique identifier: " + hx.Substring(3));
else if (hx.StartsWith("SM:")) sb.AppendLine("Sample: " + hx.Substring(3));
else sb.AppendLine("Undefined: " + hx);
}
}
return sb.ToString();
}
}
public string SQ_Details
{
get
{
StringBuilder sb = new StringBuilder();
foreach (string sx in SQ)
{
List<string> ha = B.S2T(sx);
foreach (string hx in ha)
{
if (hx.StartsWith("SN:")) sb.AppendLine("Reference sequence name: " + hx.Substring(3));
else if (hx.StartsWith("LN:")) sb.AppendLine("Reference sequence length: " + hx.Substring(3));
else if (hx.StartsWith("AH:")) sb.AppendLine("Indicates that this sequence is an alternate locus: " + hx.Substring(3));
else if (hx.StartsWith("AN:")) sb.AppendLine("Alternative reference sequence names: " + hx.Substring(3));
else if (hx.StartsWith("AS:")) sb.AppendLine("Genome assembly identifier: " + hx.Substring(3));
else if (hx.StartsWith("DS:")) sb.AppendLine("Description. UTF-8 encoding may be used.: " + hx.Substring(3));
else if (hx.StartsWith("M5:")) sb.AppendLine("MD5 checksum of the sequence in the uppercase: " + hx.Substring(3));
else if (hx.StartsWith("SP:")) sb.AppendLine("Species: " + hx.Substring(3));
else if (hx.StartsWith("TP:")) sb.AppendLine("Molecule topology: " + hx.Substring(3));
else if (hx.StartsWith("UR:")) sb.AppendLine("URI of the sequence: " + hx.Substring(3));
else sb.AppendLine("Undefined: " + hx);
}
}
return sb.ToString();
}
}
public override string ToString()
{
StringBuilder sb = new StringBuilder();
sb.AppendLine("Comment: " + CO);
sb.AppendLine(HD_Details);
sb.AppendLine(PG_Details);
sb.AppendLine(RG_Details);
sb.AppendLine(SQ_Details);
return sb.ToString();
}
}
/// <summary>
/// SAM对比项
/// E00514:173:H3C3JCCXY:4:1124:12398:67234 337 Chr00 32904 0 150M Chr09 33498107 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:0 RG:Z:J36CK1
/// E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 2469225 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:2 RG:Z:J36CK1
/// E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr16 29515410 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:4 RG:Z:J36CK1
/// E00514:173:H3C3JCCXY:4:1124:12398:67234 369 Chr00 32904 0 150M Chr17 31040767 0 TCAATTTCACTTGAAGCTTACTTGTAGTTTCAGGCTTGGTCAAGCGCGATACAAACCATGTAGTAGGAGTCCTCCAAGTCGCCAAGCTAGGGGATCTGCTGAAAGAGGTGACAGACAAGGTAAGCAATCAGAGCTCTAAGCAATCAGTCC iieiiiii`eiiiiiiiiiiiiiiieiiiiiiiieiiiiiiiiiiiiiiiiiiiiieiiiiiiiiiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieiiiiiiiii`iiieeieieeieee`` AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:136C13 YT:Z:UU NH:i:8 CC:Z:Chr10 CP:i:18604313 HI:i:6 RG:Z:J36CK1
/// E00514:173:H3C3JCCXY:4:1212:19025:24532 409 Chr00 33538 0 150M * 0 0 GATTCCAAGTGCTGACTGATTGCTCTCTTTCTCCTTGTCTTGCAGGTAAGAACAAGGCCAAAGGAAAAGACAGGGAAAAAACATGAAATGAGATACTCTTGCTTTTAACCCTGATGATATGAGATATTCTTGCTCTAGTATAGCTTGTTT ii`e`ei[iiiiiiiiiiiiie[ieeieieiiiiieiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiieee`` AS:i:-12 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:52T33T63 YT:Z:UU NH:i:20 CC:Z:Chr01 CP:i:11331871 HI:i:0 RG:Z:J36CK1
/// </summary>
public class SAM_Item
{
/// <summary>
/// 1 QNAME
/// 测序的reads的名称;
/// Query template NAME. Reads/segments having identical QNAME are regarded to come from
/// the same template.A QNAME ‘*’ indicates the information is unavailable.In a SAM file, a read may
/// occupy multiple alignment lines, when its alignment is chimeric or when multiple mappings are given.
/// </summary>
public string QNAME { get; set; } = "";
/// <summary>
/// 2 FLAG
/// 逐位FLAG组合
/// 提供了一个比对文件的信息,比对文件可以设置或取消flag,flag整数是多个flag的同时表示。
/// 表示比对的结果,由数字表示,不同的数值含义不同。见 SAM_FLAG_NAME
/// Combination of bitwise FLAGs.
/// </summary>
public string FLAG { get; set; } = "";
/// <summary>
/// 3 RNAME
/// map到参考基因组后的染色体/序列/ contig的名称;
/// Reference sequence NAME of the alignment. If @SQ header lines are present, RNAME (if not ‘*’)
/// must be present in one of the SQ-SN tag.An unmapped segment without coordinate has a ‘*’ at
/// this field.However, an unmapped segment may also have an ordinary coordinate such that it can be
/// placed at a desired position after sorting.If RNAME is ‘*’, no assumptions can be made about POS
/// and CIGAR
/// </summary>
public string RNAME { get; set; } = "";
/// <summary>
/// 4 POS
/// 比对的最左边部分的坐标;
/// 1-based leftmost mapping POSition of the first CIGAR operation that “consumes” a reference
/// base (see table below). The first base in a reference sequence has coordinate 1. POS is set as 0 for
/// an unmapped read without coordinate.If POS is 0, no assumptions can be made about RNAME and
/// CIGAR.
/// </summary>
public string POS { get; set; } = "";
/// <summary>
/// 5 MAPQ
/// 比对的映射质量;
/// MAPping Quality. It equals −10 log10 Pr{mapping position is wrong}, rounded to the nearest
/// integer.A value 255 indicates that the mapping quality is not available.
/// </summary>
public string MAPQ { get; set; } = "";
/// <summary>
/// 6 CIGAR
/// CIGAR字符串,一个数字与字母交替构成的字符串,标记了这段reads不同位置的match情况。
/// cigar中会包含数字,代表了特定match持续了多少nt;以及不同的字符,代表了不同的match情况。
/// 如36M表示它没有插入或删除。
/// </summary>
public string CIGAR { get; set; } = "";
/// <summary>
/// 7 RNEXT
/// 双末端测序中下一个reads比对的参考系列的名称,如果没有则用 " * " 表示,
/// 如果和前一个reads比对到同一个参考序列则用" = "表示;
/// Reference sequence name of the primary alignment of the NEXT read in the template. For
/// the last read, the next read is the first read in the template. If @SQ header lines are present, RNEXT (if
/// not ‘*’ or ‘=’) must be present in one of the SQ-SN tag. This field is set as ‘*’ when the information is
/// unavailable, and set as ‘=’ if RNEXT is identical RNAME. If not ‘=’ and the next read in the template
/// has one primary mapping (see also bit 0x100 in FLAG), this field is identical to RNAME at the primary
/// line of the next read. If RNEXT is ‘*’, no assumptions can be made on PNEXT and bit 0x20.
/// </summary>
public string RNEXT { get; set; } = "";
/// <summary>
/// 8 PNEXT
/// 下一个reads比对到参考序列上的位置,如果没有则用0表示;
/// 1-based Position of the primary alignment of the NEXT read in the template. Set as 0 when
/// the information is unavailable. This field equals POS at the primary line of the next read. If PNEXT
/// is 0, no assumptions can be made on RNEXT and bit 0x20.
/// </summary>
public string PNEXT { get; set; } = "";
/// <summary>
/// 9 TLEN
/// 序列模板的长度;
/// signed observed Template LENgth. For primary reads where the primary alignments of all reads
/// in the template are mapped to the same reference sequence, the absolute value of TLEN equals the
/// distance between the mapped end of the template and the mapped start of the template, inclusively
/// (i.e., end − start + 1).14 Note that mapped base is defined to be one that aligns to the reference as
/// described by CIGAR, hence excludes soft-clipped bases. The TLEN field is positive for the leftmost
/// segment of the template, negative for the rightmost, and the sign for any middle segment is undefined.
/// If segments cover the same coordinates then the choice of which is leftmost and rightmost is arbitrary,
/// but the two ends must still have differing signs. It is set as 0 for a single-segment template or when
/// the information is unavailable (e.g., when the first or last segment of a multi-segment template is
/// unmapped or when the two are mapped to different reference sequences).
/// The intention of this field is to indicate where the other end of the template has been aligned without
/// needing to read the remainder of the SAM file. Unfortunately there has been no clear consensus on
/// the definitions of the template mapped start and end. Thus the exact definitions are implementation-defined.1
/// </summary>
public string TLEN { get; set; } = "";
/// <summary>
/// 10 SEQ
/// 比对的实际顺序;
/// segment SEQuence. This field can be a ‘*’ when the sequence is not stored. If not a ‘*’, the length
/// of the sequence must equal the sum of lengths of M/I/S/=/X operations in CIGAR. An ‘=’ denotes the
/// base is identical to the reference base. No assumptions can be made on the letter cases.
/// </summary>
public string SEQ { get; set; } = "";
/// <summary>
/// 11 QUAL
/// 比对的质量字符串(fasta文件中的质量得分);
/// ASCII of base QUALity plus 33 (same as the quality string in the Sanger FASTQ format). A
/// base quality is the phred-scaled base error probability which equals −10 log10 Pr{base is wrong}. This
/// field can be a ‘*’ when quality is not stored. If not a ‘*’, SEQ must not be a ‘*’ and the length of the
/// quality string ought to equal the length of SEQ
/// </summary>
public string QUAL { get; set; } = "";
/// <summary>
/// 拼接软件基于reads之间的overlap区,拼接获得的序列称为Contig(重叠群)
/// </summary>
public string CONTIG { get; set; } = "";
/// <summary>
/// 比对的链;
/// </summary>
public string STRAND { get; set; } = "";
/// <summary>
/// 读段的长度;
/// </summary>
public string QWIDTH { get; set; } = "";
/// <summary>
/// 其他信息
/// </summary>
public List<string> EXTENSION { get; set; } = new List<string>();
public string FLAG_Details
{
get
{
if (Int32.TryParse("0" + FLAG, out var num))
{
StringBuilder sb = new StringBuilder();
if ((num & 0x0001) != 0) sb.Append(" 1:Read paired, 代表这个序列采用的是PE双端测序。");
if ((num & 0x0002) != 0) sb.Append(" 2:Read mapped in proper pair, 代表这个序列和参考序列完全匹配,没有错配和插入缺失。");
if ((num & 0x0004) != 0) sb.Append(" 4:Read unmapped, 代表这个序列没有mapping到参考序列上。");
if ((num & 0x0008) != 0) sb.Append(" 8:Mate unmapped, 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上。");
if ((num & 0x0010) != 0) sb.Append(" 16:Read reverse strand, 代表这个序列比对到参考序列的负链上。");
if ((num & 0x0020) != 0) sb.Append(" 32:Mate reverse strand, 代表这个序列对应的另一端序列比对到参考序列的负链上。");
if ((num & 0x0040) != 0) sb.Append(" 64:First in pair, 代表这个序列是R1端序列,read1。");
if ((num & 0x0080) != 0) sb.Append(" 128: Second in pair, 代表这个序列是R2端序列,read2。");
if ((num & 0x0100) != 0) sb.Append(" 256:Not primary alignment, 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的。");
if ((num & 0x0200) != 0) sb.Append(" 512:Read fails platform/vendor quality checks, 代表这个序列在QC时失败了,被过滤不掉了。");
if ((num & 0x0400) != 0) sb.Append("1024: Read is PCR or optical duplicate, 代表这个序列是PCR重复序列。");
if ((num & 0x0800) != 0) sb.Append("2048: Supplementary alignment, 代表这个序列是补充的比对。");
return sb.ToString();
}
return FLAG;
}
}
public SAM_Item(string[] array)
{
QNAME = array[0];
FLAG = array[1];
RNAME = array[2];
POS = array[3];
MAPQ = array[4];
CIGAR = array[5];
RNEXT = array[6];
PNEXT = array[7];
TLEN = array[8];
SEQ = array[9];
QUAL = array[10];
if (array.Length > 11)
{
for (int i = 11; i < array.Length; i++)
{
EXTENSION.Add(array[i]);
}
}
}
public string EXTENSION_Details
{
get
{
StringBuilder sb = new StringBuilder();
foreach (string ex in EXTENSION)
{
sb.Append("[" + ex + "] ");
string[] xa = B.S2S(ex, ':');
if (xa[0].StartsWith("X") || xa[0].StartsWith("Y") || xa[0].StartsWith("Z")) sb.AppendLine("为最终用户保留的字段(连同Y?和Z?)。Reserved fields for end users (together with Y? and Z?)");
else if (xa[0] == "AM") sb.AppendLine("其余片段的最小模板无关映射质量。The smallest template-independent mapping quality of segments in the rest.");
else if (xa[0] == "AS") sb.AppendLine("对齐器生成的对齐分数。Alignment score generated by aligner.");
else if (xa[0] == "BC") sb.AppendLine("条形码序列,任何质量分数都存储在QT标签中。Barcode sequence, with any quality scores stored in the QT tag.");
else if (xa[0] == "BQ") sb.AppendLine("偏移到基本对齐质量(BAQ),长度与读取序列相同。在第i个读取基数,BAQi=Qi−(BQi−64),其中Qi是第i个基数质量。Offset to base alignment quality (BAQ), of the same length as the read sequence. At the i-th read base, BAQi = Qi − (BQi − 64) where Qi is the i-th base quality.");
else if (xa[0] == "CC") sb.AppendLine("下一个命中的引用名称;'='对于同一条染色体。Reference name of the next hit; ‘=’ for the same chromosome");
else if (xa[0] == "CM") sb.AppendLine("编辑颜色序列和颜色参考之间的距离(另请参见NM)。Edit distance between the color sequence and the color reference (see also NM)");
else if (xa[0] == "CO") sb.AppendLine("自由文本评论。Free-text comments");
else if (xa[0] == "CP") sb.AppendLine("下一个命中的最左边坐标。Leftmost coordinate of the next hit");
else if (xa[0] == "CQ") sb.AppendLine("读取的原始链上的颜色读取质量。与QUAL编码相同;长度与CS相同。Color read quality on the original strand of the read. Same encoding as QUAL; same length as CS.");
else if (xa[0] == "CS") sb.AppendLine("读取的原始链上的颜色读取序列。必须包括底漆底座。Color read sequence on the original strand of the read. The primer base must be included.");
else if (xa[0] == "CT") sb.AppendLine("完整的读取注释标签,用于一致性注释伪特征。Complete read annotation tag, used for consensus annotation dummy features.");
else if (xa[0] == "E2") sb.AppendLine("第二个最有可能的基地呼叫。与QUAL相同的编码和长度。The 2nd most likely base calls. Same encoding and same length as QUAL.");
else if (xa[0] == "FI") sb.AppendLine("模板中段的索引。The index of segment in the template.");
else if (xa[0] == "FS") sb.AppendLine("段后缀。Segment suffix.");
else if (xa[0] == "FZ") sb.AppendLine("读取原始链上的流信号强度,存储为(uint16_t)圆形(值*100.0)。Flow signal intensities on the original strand of the read, stored as (uint16_t) round(value * 100.0).");
else if (xa[0] == "LB") sb.AppendLine("图书馆如果存在@RG,则值应与标头RG-LB标签一致。Library. Value to be consistent with the header RG-LB tag if @RG is present.");
else if (xa[0] == "H0") sb.AppendLine("完美命中次数。Number of perfect hits");
else if (xa[0] == "H1") sb.AppendLine("1-差异命中数(另请参见NM)。Number of 1-difference hits (see also NM)");
else if (xa[0] == "H2") sb.AppendLine("2差命中数。Number of 2-difference hits");
else if (xa[0] == "HI") sb.AppendLine("查询命中索引,指示对齐记录是SAM中存储的第i个对齐记录。Query hit index, indicating the alignment record is the i-th one stored in SAM");
else if (xa[0] == "IH") sb.AppendLine("SAM中存储的包含当前记录中查询的对齐数。Number of stored alignments in SAM that contains the query in the current record");
else if (xa[0] == "MC") sb.AppendLine("配对/下一段的CIGAR字符串。CIGAR string for mate/next segment");
else if (xa[0] == "MD") sb.AppendLine("位置不匹配的字符串。正则表达式:[0-9]+(([A-Z]|\^[A-Z]+)[0-9]=)*String for mismatching positions. Regex : [0-9]+(([A-Z]|\^[A-Z]+)[0-9]+)*");
else if (xa[0] == "MQ") sb.AppendLine("配对/下一段的映射质量。Mapping quality of the mate/next segment");
else if (xa[0] == "NH") sb.AppendLine("在当前记录中包含查询的已报告路线数。Number of reported alignments that contains the query in the current record");
else if (xa[0] == "NM") sb.AppendLine("编辑到参照的距离,包括不明确的基准,但不包括剪裁。Edit distance to the reference, including ambiguous bases but excluding clipping");
else if (xa[0] == "OQ") sb.AppendLine("原始基准质量(通常在重新校准之前)。与QUAL编码相同。Original base quality (usually before recalibration). Same encoding as QUAL.");
else if (xa[0] == "OP") sb.AppendLine("原始映射位置(通常在重新对齐之前)。Original mapping position (usually before realignment)");
else if (xa[0] == "OC") sb.AppendLine("原始CIGAR(通常在重新调整之前)。Original CIGAR (usually before realignment)");
else if (xa[0] == "PG") sb.AppendLine("程序如果存在@PG,则值与标头PG-ID标记匹配。Program. Value matches the header PG-ID tag if @PG is present.");
else if (xa[0] == "PQ") sb.AppendLine("模板的Phred似然性,条件是映射都是正确的。Phred likelihood of the template, conditional on both the mapping being correct");
else if (xa[0] == "PT") sb.AppendLine("填充读取序列部分的读取注释11。Read annotations for parts of the padded read sequence11");
else if (xa[0] == "PU") sb.AppendLine("平台单元。如果存在@RG,则值应与标头RG-PU标签一致。Platform unit. Value to be consistent with the header RG-PU tag if @RG is present.");
else if (xa[0] == "QT") sb.AppendLine("BC(或RT)标签中条形码序列的Phred质量。与QUAL编码相同。Phred quality of the barcode sequence in the BC (or RT) tag. Same encoding as QUAL.");
else if (xa[0] == "Q2") sb.AppendLine("R2标签中的配对/下一个片段序列的Phred质量。与QUAL编码相同。Phred quality of the mate/next segment sequence in the R2 tag. Same encoding as QUAL.");
else if (xa[0] == "R2") sb.AppendLine("模板中的配合/下一个分段的顺序。Sequence of the mate/next segment in the template.");
else if (xa[0] == "RG") sb.AppendLine("读取组。如果标头中存在@RG,则值与标头RG-ID标记匹配。Read group. Value matches the header RG-ID tag if @RG is present in the header.");
else if (xa[0] == "RT") sb.AppendLine("Sanger最初使用的BC标签的弃用替代品。Deprecated alternative to BC tag originally used at Sanger.");
else if (xa[0] == "SA") sb.AppendLine("嵌合比对中的其他规范比对,格式为分号分隔的列表:(rname,pos,strand,CIGAR,mapQ,NM;)+。列表中的每个元素表示嵌合排列的一部分。传统上,在辅助线处,第一个元素指向主线。Other canonical alignments in a chimeric alignment, formatted as a semicolon-delimited list: (rname, pos, strand, CIGAR, mapQ, NM;)+. Each element in the list represents a part of the chimeric alignment. Conventionally, at a supplementary line, the first element points to the primary line.");
else if (xa[0] == "SM") sb.AppendLine("与模板无关的映射质量。Template-independent mapping quality");
else if (xa[0] == "TC") sb.AppendLine("模板中的分段数。The number of segments in the template.");
else if (xa[0] == "U2") sb.AppendLine("第二次调用出错的概率以最佳调用出错为条件。与QUAL编码相同。Phred probility of the 2nd call being wrong conditional on the best being wrong. The same encoding as QUAL.");
else if (xa[0] == "UQ") sb.AppendLine("分段的Phred似然性,条件是映射正确。Phred likelihood of the segment, conditional on the mapping being correct");
else sb.AppendLine("未知。Unknowed.");
}
return sb.ToString();
}
}
public override string ToString()
{
StringBuilder sb = new StringBuilder();
sb.AppendLine("1 QNAME");
sb.AppendLine(" 测序的reads的名称。");
sb.AppendLine(" Query template NAME. Reads/segments having identical QNAME are regarded to come from");
sb.AppendLine(" the same template.A QNAME ‘*’ indicates the information is unavailable.In a SAM file, a read may");
sb.AppendLine(" occupy multiple alignment lines, when its alignment is chimeric or when multiple mappings are given.");
sb.AppendLine(" " + QNAME);
sb.AppendLine("");
sb.AppendLine("2 FLAG");
sb.AppendLine(" Combination of bitwise FLAGs.");
sb.AppendLine(" 逐位FLAG组合");
sb.AppendLine(" 提供了一个比对文件的信息,比对文件可以设置或取消flag,flag整数是多个flag的同时表示。");
sb.AppendLine(" 表示比对的结果,由数字表示,不同的数值含义不同。");
sb.AppendLine(" " + FLAG);
sb.AppendLine(" " + FLAG_Details);
sb.AppendLine("");
sb.AppendLine("3 RNAME");
sb.AppendLine(" map到参考基因组后的染色体/序列/ contig的名称;");
sb.AppendLine(" Reference sequence NAME of the alignment. If @SQ header lines are present, RNAME (if not ‘*’)");
sb.AppendLine(" must be present in one of the SQ-SN tag.An unmapped segment without coordinate has a ‘*’ at");
sb.AppendLine(" this field.However, an unmapped segment may also have an ordinary coordinate such that it can be");
sb.AppendLine(" placed at a desired position after sorting.If RNAME is ‘*’, no assumptions can be made about POS");
sb.AppendLine(" and CIGAR");
sb.AppendLine(" " + RNAME);
sb.AppendLine("");
sb.AppendLine("4 POS");
sb.AppendLine(" 比对的最左边部分的坐标;");
sb.AppendLine(" 1-based leftmost mapping POSition of the first CIGAR operation that “consumes” a reference");
sb.AppendLine(" base (see table below). The first base in a reference sequence has coordinate 1. POS is set as 0 for");
sb.AppendLine(" an unmapped read without coordinate.If POS is 0, no assumptions can be made about RNAME and");
sb.AppendLine(" CIGAR.");
sb.AppendLine(" " + POS);
sb.AppendLine("");
sb.AppendLine("5 MAPQ");
sb.AppendLine(" 比对的映射质量;");
sb.AppendLine(" MAPping Quality. It equals −10 log10 Pr{mapping position is wrong}, rounded to the nearest");
sb.AppendLine(" integer.A value 255 indicates that the mapping quality is not available.");
sb.AppendLine(" " + MAPQ);
sb.AppendLine("");
sb.AppendLine("6 CIGAR");
sb.AppendLine(" CIGAR字符串,一个数字与字母交替构成的字符串,标记了这段reads不同位置的match情况。");
sb.AppendLine(" cigar中会包含数字,代表了特定match持续了多少nt;以及不同的字符,代表了不同的match情况。");
sb.AppendLine(" 如36M表示它没有插入或删除。");
sb.AppendLine(" " + CIGAR);
sb.AppendLine("");
sb.AppendLine("7 RNEXT");
sb.AppendLine(" 双末端测序中下一个reads比对的参考系列的名称,如果没有则用 * 表示,");
sb.AppendLine(" 如果和前一个reads比对到同一个参考序列则用 = 表示;");
sb.AppendLine(" Reference sequence name of the primary alignment of the NEXT read in the template. For");
sb.AppendLine(" the last read, the next read is the first read in the template. If @SQ header lines are present, RNEXT (if");
sb.AppendLine(" not ‘*’ or ‘=’) must be present in one of the SQ-SN tag. This field is set as ‘*’ when the information is");
sb.AppendLine(" unavailable, and set as ‘=’ if RNEXT is identical RNAME. If not ‘=’ and the next read in the template");
sb.AppendLine(" has one primary mapping (see also bit 0x100 in FLAG), this field is identical to RNAME at the primary");
sb.AppendLine(" line of the next read. If RNEXT is ‘*’, no assumptions can be made on PNEXT and bit 0x20.");
sb.AppendLine(" " + RNEXT);
sb.AppendLine("");
sb.AppendLine("8 PNEXT ");
sb.AppendLine(" 下一个reads比对到参考序列上的位置,如果没有则用0表示;");
sb.AppendLine(" 1-based Position of the primary alignment of the NEXT read in the template. Set as 0 when");
sb.AppendLine(" the information is unavailable. This field equals POS at the primary line of the next read. If PNEXT");
sb.AppendLine(" is 0, no assumptions can be made on RNEXT and bit 0x20.");
sb.AppendLine(" " + PNEXT);
sb.AppendLine("");
sb.AppendLine("9 TLEN");
sb.AppendLine(" 序列模板的长度;");
sb.AppendLine(" signed observed Template LENgth. For primary reads where the primary alignments of all reads");
sb.AppendLine(" in the template are mapped to the same reference sequence, the absolute value of TLEN equals the");
sb.AppendLine(" distance between the mapped end of the template and the mapped start of the template, inclusively");
sb.AppendLine(" (i.e., end − start + 1).14 Note that mapped base is defined to be one that aligns to the reference as");
sb.AppendLine(" described by CIGAR, hence excludes soft-clipped bases. The TLEN field is positive for the leftmost");
sb.AppendLine(" segment of the template, negative for the rightmost, and the sign for any middle segment is undefined.");
sb.AppendLine(" If segments cover the same coordinates then the choice of which is leftmost and rightmost is arbitrary,");
sb.AppendLine(" but the two ends must still have differing signs. It is set as 0 for a single-segment template or when");
sb.AppendLine(" the information is unavailable (e.g., when the first or last segment of a multi-segment template is");
sb.AppendLine(" unmapped or when the two are mapped to different reference sequences).");
sb.AppendLine(" The intention of this field is to indicate where the other end of the template has been aligned without");
sb.AppendLine(" needing to read the remainder of the SAM file. Unfortunately there has been no clear consensus on");
sb.AppendLine(" the definitions of the template mapped start and end. Thus the exact definitions are implementation-defined.1");
sb.AppendLine(" " + TLEN);
sb.AppendLine("");
sb.AppendLine("10 SEQ");
sb.AppendLine(" 比对的实际顺序;");
sb.AppendLine(" segment SEQuence. This field can be a ‘*’ when the sequence is not stored. If not a ‘*’, the length");
sb.AppendLine(" of the sequence must equal the sum of lengths of M/I/S/=/X operations in CIGAR. An ‘=’ denotes the");
sb.AppendLine(" base is identical to the reference base. No assumptions can be made on the letter cases.");
sb.AppendLine(" " + SEQ);
sb.AppendLine("");
sb.AppendLine("11 QUAL");
sb.AppendLine(" 比对的质量字符串(fasta文件中的质量得分)");
sb.AppendLine(" ASCII of base QUALity plus 33 (same as the quality string in the Sanger FASTQ format). ");
sb.AppendLine(" A base quality is the phred-scaled base error probability which equals −10 log10 Pr{base is wrong}. ");
sb.AppendLine(" This field can be a ‘*’ when quality is not stored. If not a ‘*’, SEQ must not be a ‘*’ and the length of the");
sb.AppendLine(" quality string ought to equal the length of SEQ");
sb.AppendLine(" " + QUAL);
sb.AppendLine("");
sb.AppendLine("MORE++");
sb.AppendLine(EXTENSION_Details);
return sb.ToString();
}
}
public class SAM_File
{
/// <summary>
/// HEAD部分
/// </summary>
public SAM_Head Head { get; set; } = new SAM_Head();
/// <summary>
/// 对比项集合
/// </summary>
public List<SAM_Item> Items { get; set; } = new List<SAM_Item>();
public SAM_File(string buf)
{
try
{
string[] xlines = buf.Split(new char[] { '
', '
' }, StringSplitOptions.RemoveEmptyEntries);
for (int i = 0; i < xlines.Length; i++)
{
if (xlines[i].StartsWith("@"))
{
if (xlines[i].StartsWith("@HD")) Head.HD = xlines[i];
else if (xlines[i].StartsWith("@SQ")) Head.SQ.Add(xlines[i]);
else if (xlines[i].StartsWith("@PG")) Head.PG = xlines[i];
else throw new Exception("UNKONW SAM Head " + xlines[i]);
}
else
{
string[] xa = xlines[i].Split(new char[] { ' ' }, StringSplitOptions.RemoveEmptyEntries);
if (xa.Length >= 11)
{
Items.Add(new SAM_Item(xa)); ;
}
else
{
throw new Exception("UNKONW SAM Item " + xlines[i]);
}
}
}
}
catch (Exception ex)
{
throw new Exception("SAM() ERROR:" + ex.Message);
}
}
public void FromFile(string filename)
{
try
{
string buf = File.ReadAllText(filename);
new SAM_File(buf);
}
catch (Exception ex)
{
throw new Exception("FromFile ERROR:" + ex.Message);
}
}
}
}




站长推荐
- QT多线程的5种用法,通过使用线程解决UI主界面的耗时操作代码,防止界面卡死。
 QT多线程的5种用法,通过使用线程解决UI主界面的耗时操作代码,防止界面卡死。...
QT多线程的5种用法,通过使用线程解决UI主界面的耗时操作代码,防止界面卡死。... - U8W/U8W-Mini使用与常见问题解决
 U8W/U8W-Mini使用与常见问题解决
U8W/U8W-Mini使用与常见问题解决 - stm32使用HAL库配置串口中断收发数据(保姆级教程)
 stm32使用HAL库配置串口中断收发数据(保姆级教程)
stm32使用HAL库配置串口中断收发数据(保姆级教程) - 分享几个国内免费的ChatGPT镜像网址(亲测有效)
 分享几个国内免费的ChatGPT镜像网址(亲测有效)
分享几个国内免费的ChatGPT镜像网址(亲测有效) - Allegro16.6差分等长设置及走线总结
 Allegro16.6差分等长设置及走线总结
Allegro16.6差分等长设置及走线总结